modPhred: Documentation
What is modPhred?
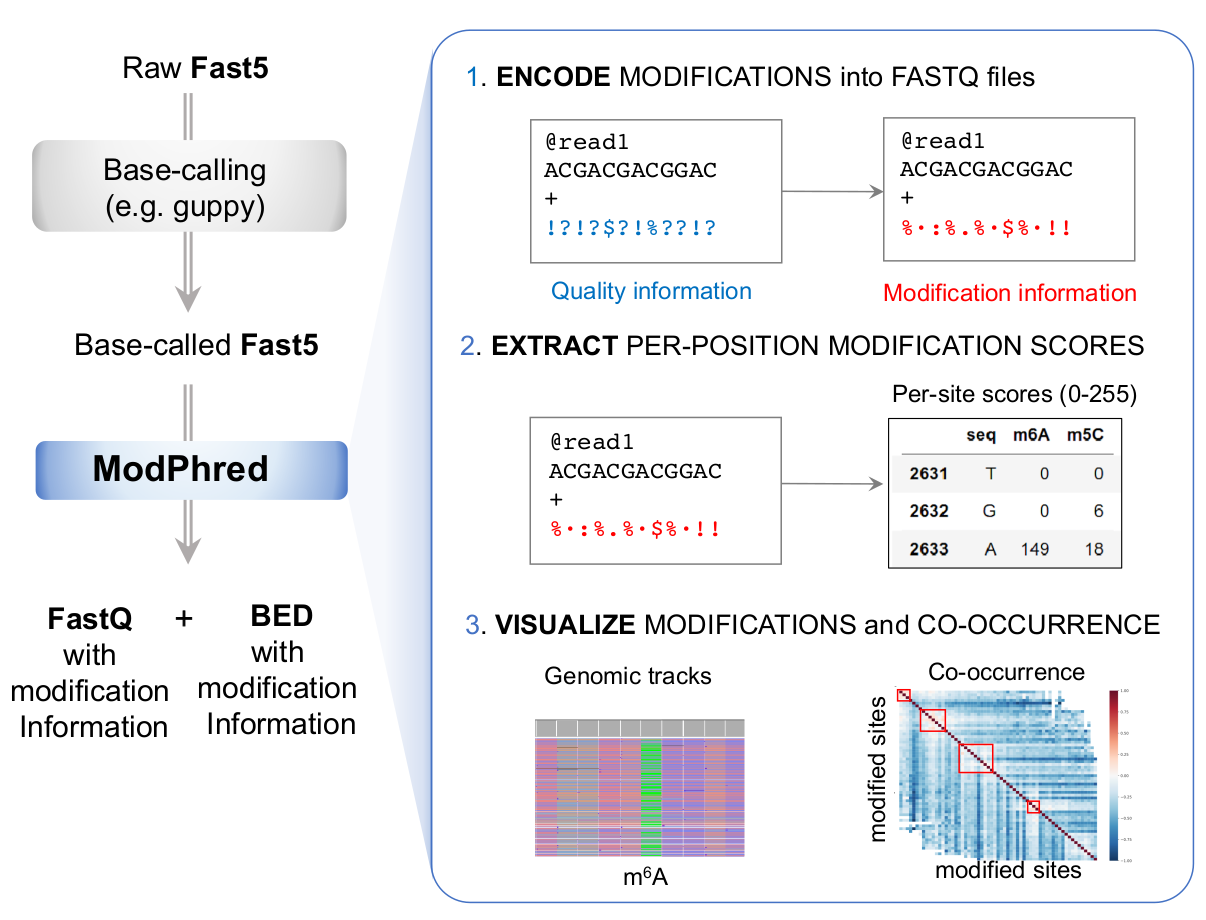
modPhred is a pipeline for detection, annotation and visualisation of DNA/RNA modifications. The pipeline consists of four steps / modules:
modEncode: encoding modification probabilities in FastQ (mod_encode.py)
modAlign: build alignments keepind modification information in BAMs (mod_align.py)
modReport: extraction of RNA modification information (bedGraph) and QC reports (mod_report.py)
modAnalysis:
plotting QC stats, pairplots & venn diagrams (mod_plot.py),
co-occurrence of modifications(mod_correlation.py)
per-read clustering based on modification profiles (mod_cluster.py)
All these scripts can be run separately or as a pipeline by executing modPhred/run.
You can find more the details in the Methods section.
What do I need to run ModPhred?
To run modPhred, you will need:
reference sequence (FastA)
raw ONT data (Fast5)
modification-aware guppy_basecaller model.
Currently, there is only one modification-aware model distributed together with guppy. You can find more experimental models or you can train your own models using taiyaki.
Why use modPhred?
Cause why not! And seriously, it is:
free (MIT licensed) & fast (a few times faster than other tools)
easy-to-use & versatile: will do all for you with just one command (or at least encoding of modifications in FastQ, alignments, detection of modified positions, QC & plotting…)
powerfull & space-optimised: it stores the modification status inside FastQ/BAM
no external files/DBs needed
you can visualise modification status of all bases of all reads in your favourite genome browser ie IGV
you can remove all basecalled Fast5 files to save disk space or skip separate basecalling step entirely thanks to on-the-fly basecalling!
visually attractive: it produces nice plots (or at least not so bad so far… still working on it;) )
Getting help
If you have any questions, issues or doubts, first please get familiar with our documentation. This should address most common questions/problems. Then, have a look at issues users reported so far. If you don’t find the solution, please open a new one.
What does modPhred stand for?
The tool stores base modification status (probability of base having various types of modifications) encoded inside FastQ/BAM file instead of base quality (also called Phred scores), thus mod (for modification) & Phred (for base quality).. Or something like that :P
Initially, this tool was called Pszczyna, pronounced ˈpʂt͡ʂɨna, but since almost no one could pronounce or memorise it, we came up with much easier, yet so much less sexy name… There are numerous more interesting words that Pszczyna in Polish. For more information, check Wikipedia.
Citation
If you find this work useful, please cite:
Pryszcz LP and Novoa EM (2022) ModPhred: an integrative toolkit for the analysis and storage of nanopore sequencing DNA and RNA modification data. Bioinformatics, 38:257-260.
Please, consider citing dependencies as well.